2019年3月13日,南京大学科研团队再登Nature杂志。该校戈惠明、谭仁祥和梁勇研究团队首次鉴定出能够催化[6+4]环加成反应的一类酶家族,相关成果“Enzyme-catalysed [6+4] cycloadditions in the biosynthesis of natural products”在线发表在Nature杂志上。
(南京大学助理研究员张博博士、以及博士研究生王凯标、王文和汪欣为该论文共同第一作者。戈惠明、谭仁祥、梁勇以及加州大学洛杉矶分校的Kendall N. Houk教授为共同通讯作者。)
周环反应是一类在反应过程中形成环状过渡态的协同反应,比如著名的[4+2]环加成反应(Diels-Alder反应)、Cope-重排反应、Claisen-重排反应等,这些反应在有机合成中有着广泛的应用。有意思的是,在自然界中也观察到了可催化这些著名周环反应的酶。早在1965年,Woodward和Hoffmann两位诺贝尔化学奖获得者就预测了[6+4]或其它高阶环加成反应可以发生,随后在有机合成中确实观察到了[6+4]反应,但自然界中是否存在着可催化[6+4]反应的酶仍然是个谜。

该团队在前期工作中,从海洋放线菌中发现了一个具有抗幽门螺旋杆菌活性的新颖大环内酯化合物命名为streptoseomycin,根据其结构推断,它在微生物体内的合成过程中可能涉及[4+2]环加成反应。
为鉴定此过程,他们比较分析了streptoseomycin以及其结构类似物nargenicin的生物合成基因簇,推测一个未知功能的同源蛋白StmD、NgnD很有可能分别负责streptoseomycin和nargenicin中的环化反应。然而,敲除stmD基因并未得到预料中的环化产物前体3,而得到了线性化合物4和5,这很可能是化合物3环内双键较多,张力较大,自发水解开环所致。研究人员设计了一个在微生物体内验证StmD功能的实验,首先敲除了基因簇上大部分基因,仅留下聚酮合成酶基因,此突变株只产生化合物4;当将stmD基因回补回去时,得到了化合物6-8,其中化合物7不稳定,可经Cope重排转化成6。该结果表明,聚酮合成酶的直接产物是3,若无下游其它酶存在时,3将发生水解开环而生成4;若再引入StmD,则催化发生周环反应,奇怪的是,该酶不仅催化了[4+2]反应,也催化了[6+4]反应。
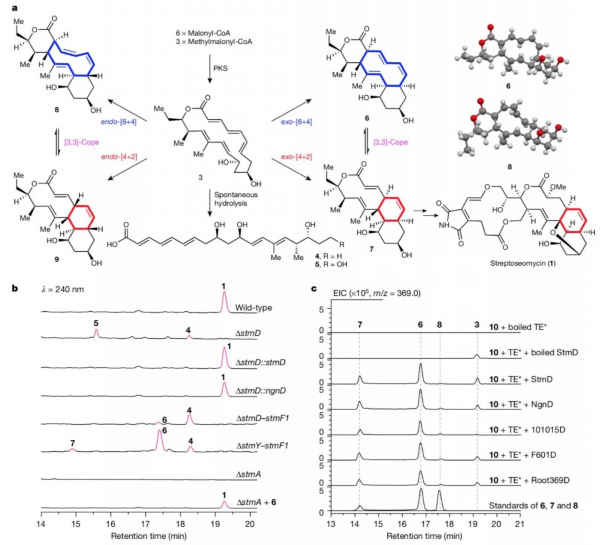
天然产物生物合成中的环加成。图片来源:Nature
随后,研究人员设计了两步串联的酶反应在体外来验证周环酶的催化功能。首先,通过聚酮合成酶上的硫酯酶结构域催化链状化合物4-SNAC环化形成3,当同时加入StmD或NgnD时,反应体系中观察到了化合物6-8的生成,确证StmD或NgnD可同时催化[4+2]和[6+4]反应。此外研究人员以StmD为探针从公共基因组数据库中,挖掘鉴定出了另外三个可催化此反应的酶。
为了更好地理解[6+4]、[4+2]环加成反应和[3,3]-Cope重排反应之间的相互关系,研究人员通过密度泛函理论(DFT)计算了反应的热力学和动力学。有趣的是,底物3越过单一过渡态后可歧化成两个方向,分别生成[6+4]、[4+2]反应产物,由于[6+4]化合物在热力学上更稳定,[4+2]产物可再经Cope重排自发转化成[6+4]产物。此理论计算结果与实验观察结果相吻合,进一步证明了之前的推断。
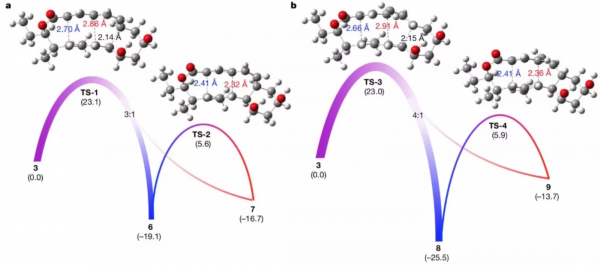
反应热力学和动力学的DFT计算。图片来源:Nature
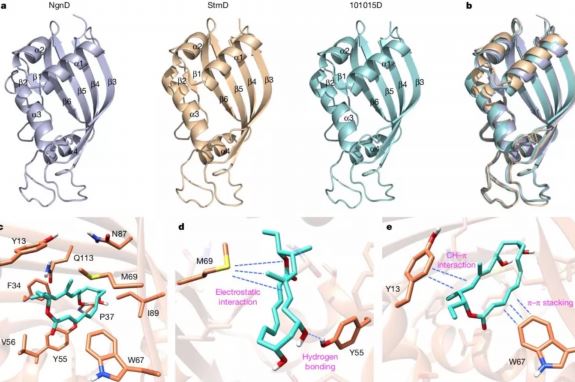
最后,研究者获得了StmD、NgnD和101015D三个蛋白的晶体来进一步研究该反应的催化机理。通过分子对接、计算机模拟和点突变实验,阐明了此类新型环加成酶的反应机制。M69中富含电子的硫原子会特定的指向过渡态中部分带正电的双烯部分,从而产生有利的静电作用;同时反应物中部分带负电的三烯酯会与W67上的芳香环产生π–π堆叠相互作用,来稳定过渡态并加速整个反应的进行。此外,Y55和Y13也会通过分子间氢键和CH–π相互作用来催化反应。
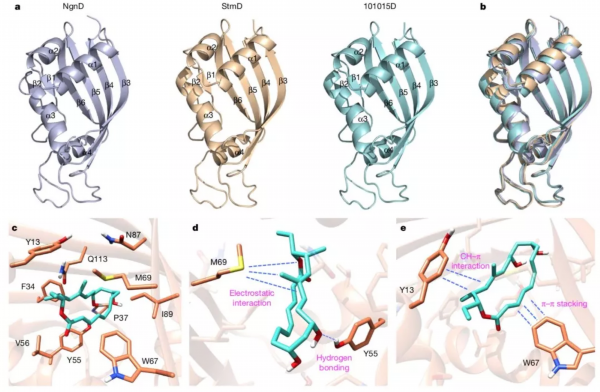
酶和催化活性位点的结构分析。图片来源:Nature
综上所述,研究人员巧妙设计实验,通过体内敲除基因、体外酶催化反应、量子化学计算、分子动力学模拟以及蛋白晶体的研究等,表征了首例可催化[6+4]/[4+2]环加成反应的酶。这类酶的发现将进一步拓展人们对周环反应酶的认识,启发科学家们将来利用和改造周环反应酶来实现有价值的分子转化。
(来源:X一MOL资讯)





